Chemically Modified Single-Stranded DNA Donors Enable Efficient mRNA Gene Editing-Mediated Knock-In in Human iPS Cells
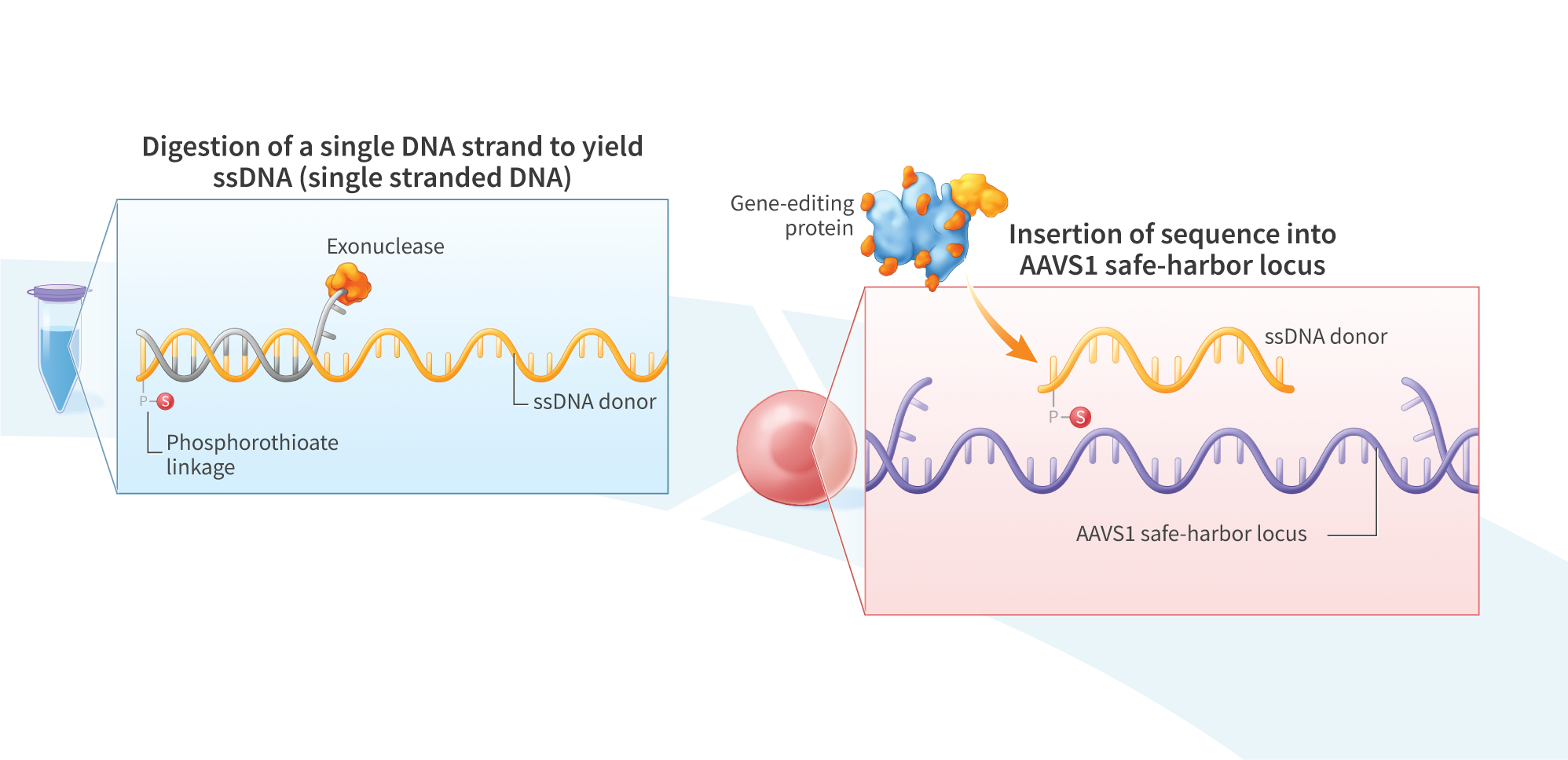
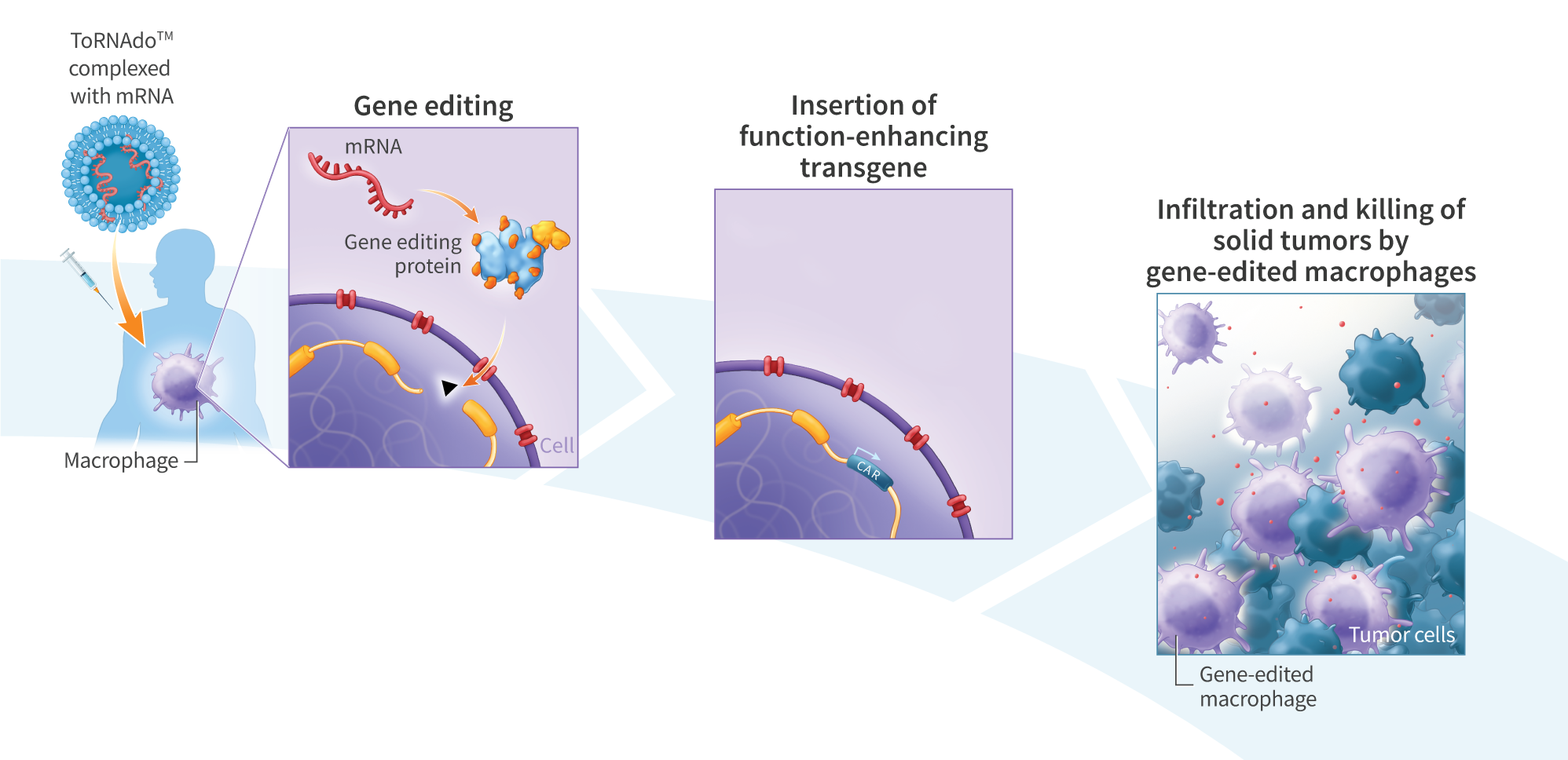
Genome-editing technology provides a means of modifying genes in living cells, and is being explored for the development of therapies to treat cancer and a variety of genetic disorders. Gene-editing proteins can be used to create single- and double-strand breaks at specific genomic sites for knocking out a gene or, when combined with an exogenous DNA donor, knock-in of a defined sequence. Compared with double-stranded DNA (dsDNA), single-stranded DNA (ssDNA) exhibits lower toxicity and is less prone to random genomic integration, making it an attractive form of DNA donor for gene knock-in. However, current methods of synthesizing ssDNA, including enzymatic digestion, asymmetric PCR, and chemical synthesis, suffer from low yields, contamination with residual dsDNA, length limitations, and high cost. Here, we present an enzymatic approach for producing long (>3kb) and concentrated (>1pg/uL) ssDNA suitable for generation of knock-in lines of human induced pluripotent stem (hiPS) cells. We PCR-amplified dsDNA from plasmid templates using 5’-modified primers incorporating a 5’ phosphate on the strand intended for digestion and a single 5’phosphorothioate linkage on the protein-encoding strand to protect against digestion. The resulting PCR products were treated with lambda exonuclease, which preferentially digests strands with a 5’phosphate group, to yield a single-stranded product. To minimize degradation of the ssDNA, we included in the reaction a short, double-stranded oligonucleotide (dsDecoy), for which lambda exonuclease has higher affinity than ssDNA. Reactions were further treated with the less processive T7 exonuclease to eliminate residual dsDNA not digested by lambda exonuclease, yielding concentrated (>1pug/uL) ssDNA. Gel electrophoresis analysis of five products ranging from 1.4kbto 3.3kb revealed a single, sharp band in the region of interest. Four of the ssDNA products contained less than 0.3% residual dSDNA by mass, as determined by gel electrophoresis using a double-stranded standard of known concentration. The fifth, 3.3kb ssDNA product contained 1.1% dsDNA by mass. To test their utility as knock-in donors, ssDNA products were co-electroporated into hiPS cells with mRNA encoding UltraSlice gene-editing proteins targeting the AAVSI safe harbor locus. Insertion efficiencies were 67.8% for a 1.2kb donor, 8.6% for a 2kb donor encoding a ROR1 CAR, and 2.7% for a 2.8kb donor encoding green fluorescent protein. In summary, we demonstrate a method for high-yield synthesis of ssDNA suitable for cellular applications, including mRNA gene editing-mediated knock-in in iPS$ cells. Overall, this platform may prove useful in the development of gene-editing therapeutics.